SPICA: Surface Property fItting Coarse grAined model
Tutorialprotein
Step 3. Convert PDB file to LAMMPS input file
In the previous step, we prepared the topology file (protein.cg.top) needed for generating LAMMPS input files for CG-MD simulations. As seen in tutorial for lipid membrane systems, to convert CG PDB file to LAMMPS data file and gain LAMMPS parameter file from database file of SPICA FF for protein, we use a command of spica-tools setup_lmp.download Database file for SPICA protein
Because the PDB file generated with CHARMM-GUI does not have the simulation box information, we need to add the information manually using the "sed" unix command like:
$ sed -i 1i"CRYST1 69.000 69.000 90.000 90.00 90.00 90.00 P 1 1" system.cg.pdb
The simulation box size is written in the "step5_assembly.str" file included in the generated CHARMM-GUI top directory.
The command line to excute the "setup_lmp" is:
$ cg_spica setup_lmp protein.cg.top 1 DOPC.top 128 WAT.top 2134 CLA.top 4 spica_protein_db.prm system.cg.pdb
where the "DOPC.top", "WAT.top", and "CLA.top" contain the topology of DOPC, water, and chloride ion, respectively. These topology files are included in the force field file (Check "./topology/*.top"). "spica_protein_db.prm" is the force field file we provide. "system.cg.pdb" is the PDB file contains the intial CG coordinates generated in Step 1 of this tutorial. One should be careful with the order of arguments. Especially, molecule topology and number should be the same as in the PDB file.
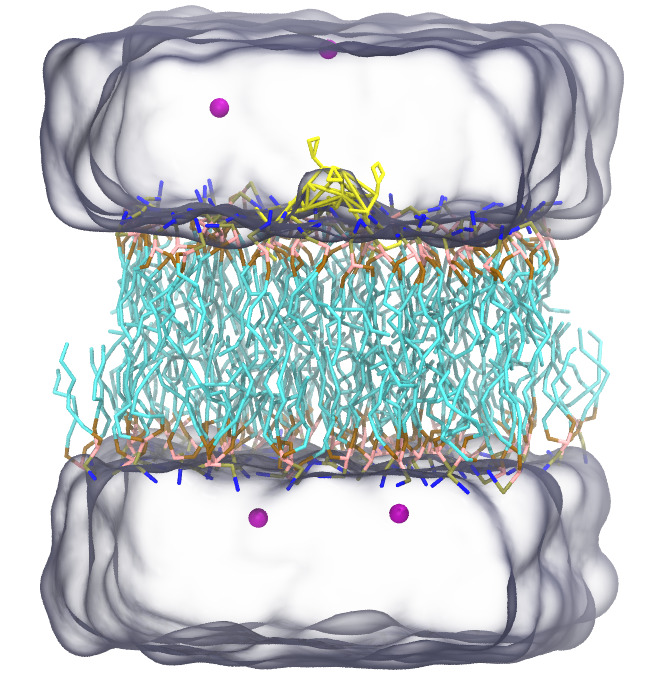
If the conversion is successful, setup_lammps will generate two output files, "DATA.FILE" and "PARM.FILE". The input script will read these files. The contents of "DATA.FILE" can be visualized on VMD with Topotools.
% package require topotools
% topo readlammpsdata DATA.FILE
% topo readlammpsdata DATA.FILE
In addition, the "setup_lmp" command gives PSF file of the target system. We can visualize the system using the output PSF file (out.psf) and input PDB file (system.cg.pdb):
$ vmd out.psf system.cg.pdb
| Previous | Top | Next |
SPICA Force Field
Research Institute for Interdisciplinary Science (RIIS)
Okayama University