SPICA: Surface Property fItting Coarse grAined model
Force Field
SPICA Force Field
We describe the molecule using several different interaction functions in this force field. This is mostly the same as used in the SDK model, though a torsion potential term is included for cholesterol model. For intramolecular interaction, we consider harmonic bond stretching and angle bending potentials for 1-2 and 1-2-3 bonded pairs, while the pairs separated by more than two bonds interact via the nonbonded force.
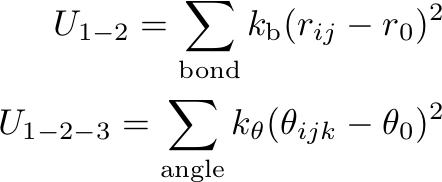
where kb and kθ are the force constants and r0 and θ0 are the distance and angle at the minimum energy, respectively.
We have also a correction term for 1-3 interaction, which is the same as the nonbonded (Lennard-Jones, LJ) interaction but being truncated-shifted to get zero at r = rs.
rs is chosen at the energy minimum distance, rmin, of LJ function. Thus, this correction term is just a short-range interaction and does not work always but effectively prevents unwilling bent structure.
Torsion potential is not typically considered, though is applied to only selected pairs in the sterol tail;
Nonbond interaction is described by LJ functions and Coulomb interaction. Two LJ functions are used, depending on the pair of particles;
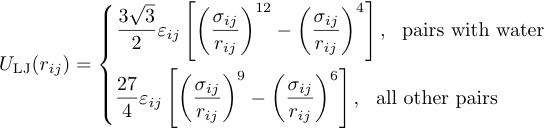
These interaction is treated by a plane-cutoff scheme with the cutoff length of 1.5 nm. The coulomb interaction
is considered only between ionic segments and should be calculated by a non-cutoff scheme like particle mesh Ewald or P3M. We set the εr=80 to incorporate the screenning effect of water implicitly in this model. Note that, in the actual calculation using a software (LAMMPS, Gromacs, MPDyn), we do not set εr=80 explicitly, instead the distributed topology file describe the point charges on ionic particles with a scaled value (scaled by 1/sqrt(80)). The point charges (q=+1 or -1) are given as (q=+0.1118 or -0.1118) in the toppology files. So the computation should be done with εr=1.
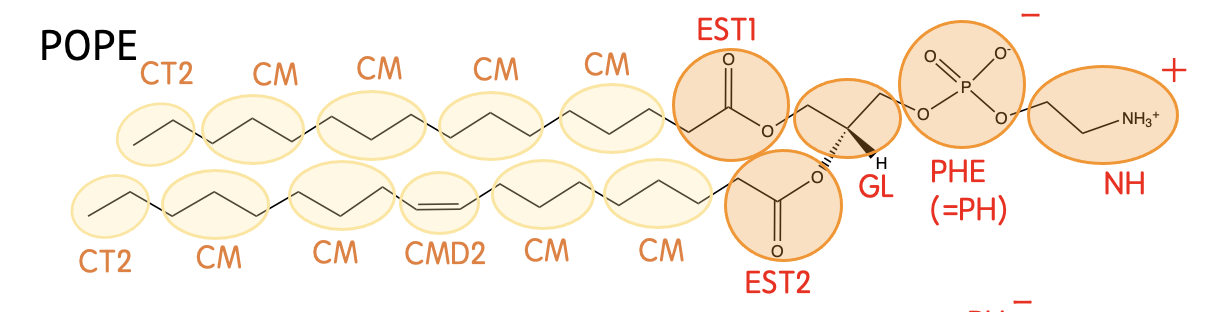
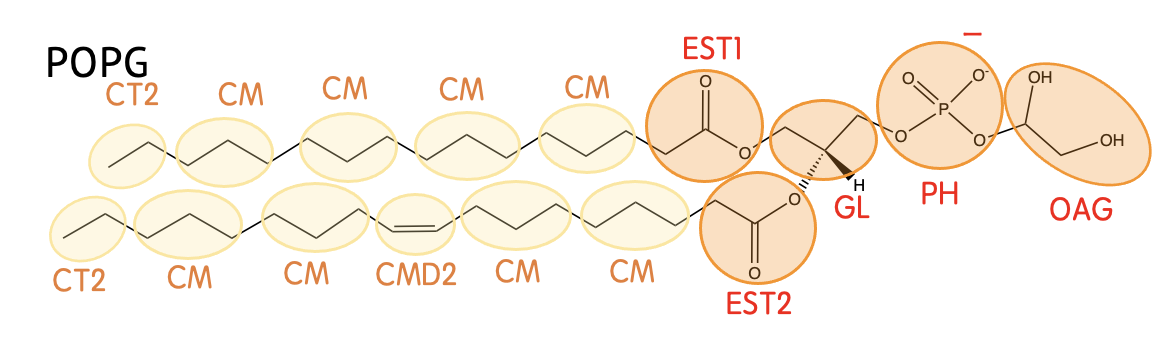
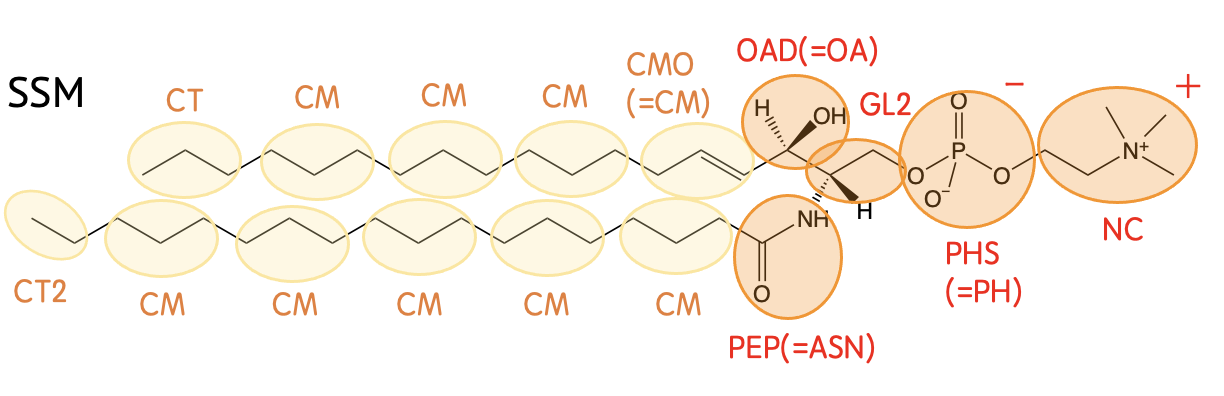
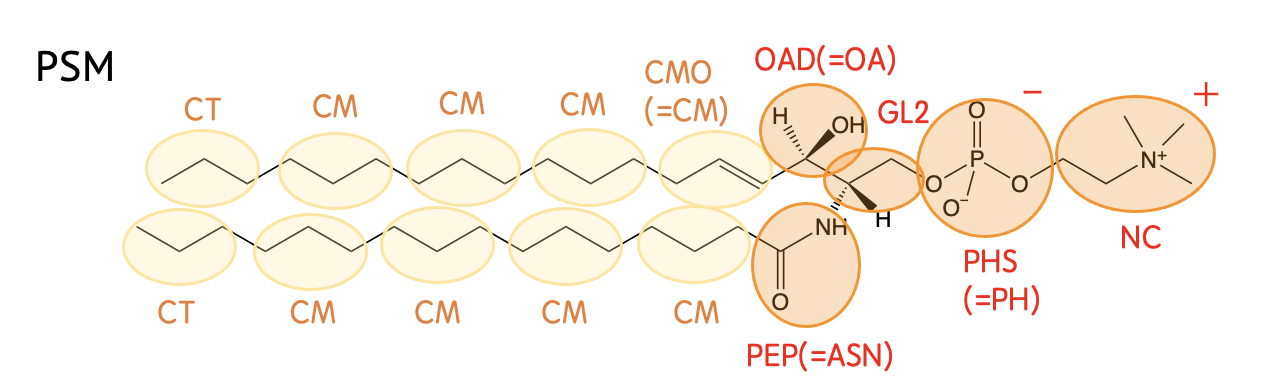
Mapping
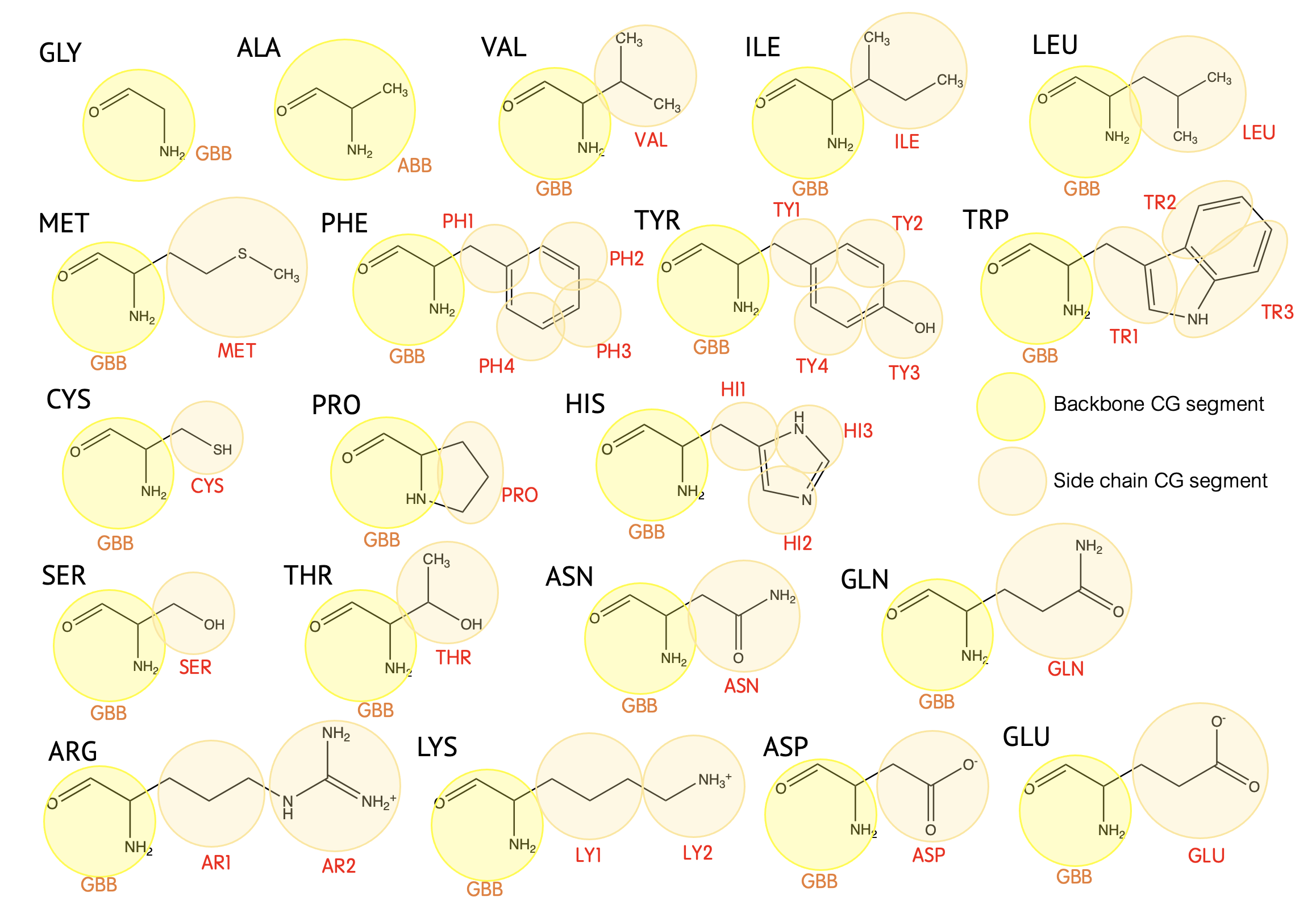 |
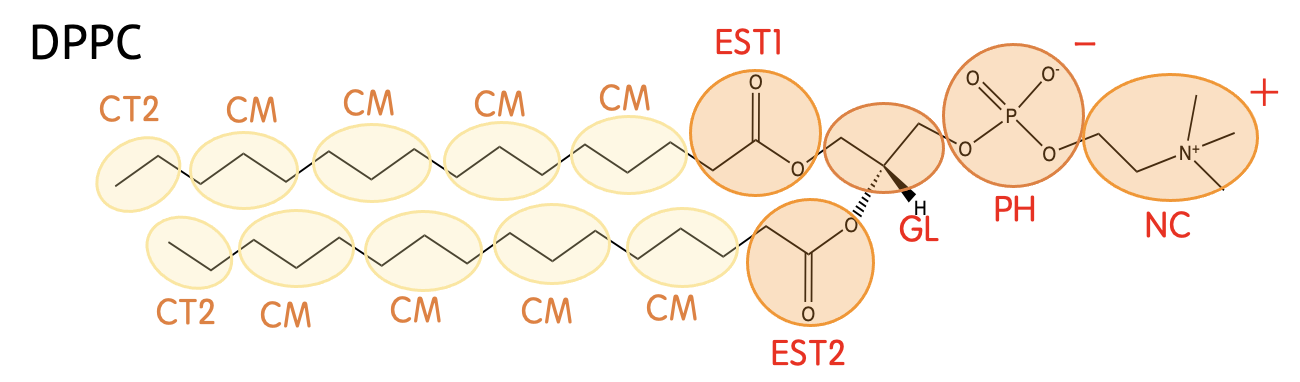 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
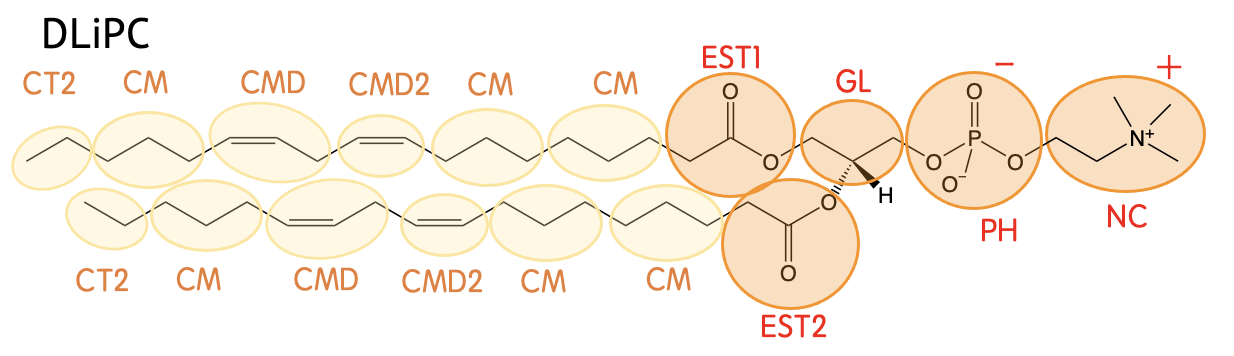
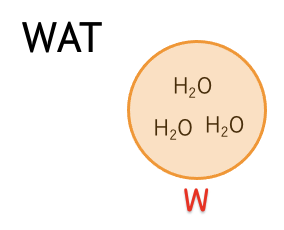
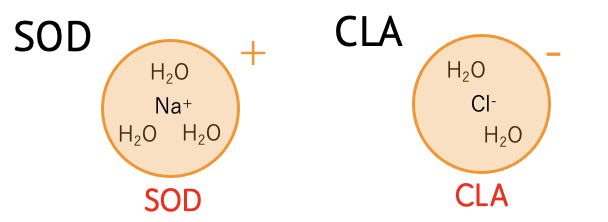
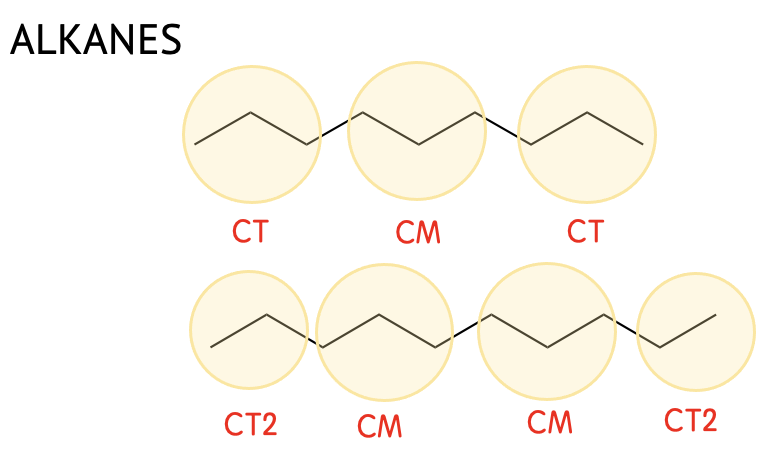
Definision
| CG-Name | All-atom |
| W | (H2O)3 |
| SOD | (H2O)3 Na+ (+1) |
| CLA | (H2O)2 Cl- (-1) |
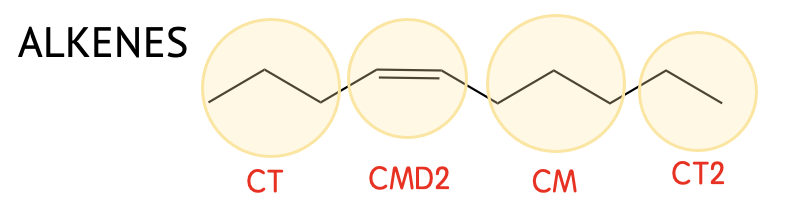
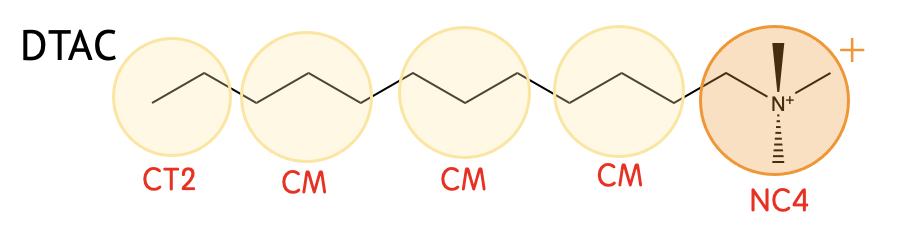
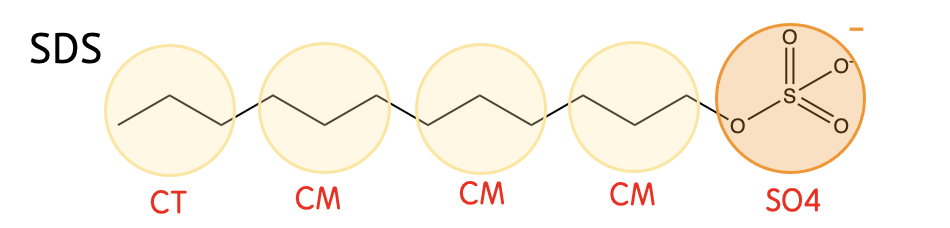
| CT | CH3CH2CH2- |
| CM | -CH2CH2CH2- |
| CT2 | CH3CH2- |
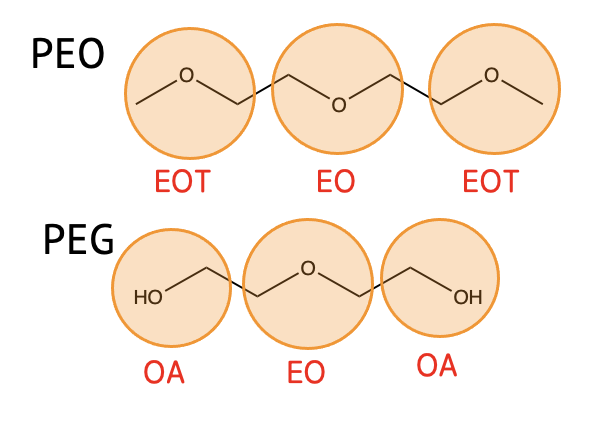
| EOT | CH3-O-CH2- |
| EO | -CH2-O-CH2- |
| OA | HOCH2- |
| NC | -CH2CH2-N-(CH3)3 (+1) |
| NH | -CH2CH2-NH3 (+1) |
| PH | -PO4- (-1) |
| PHE | -PO4- (-1) (for PE headgroup) |
| GL | -CH2CH-CH2- |
| EST1 | -CH2CO2- (in the sn-2 chain) |
| EST2 | -CH2CO2- (in the sn-1 chain) |
| CMD2 | -HC=CH- (cis) |
| SO4 | -SO4 (-1) |
| NC4 | (CH3)3N-CH2- (+1) |
| CM2 | -CH2CH2- |
| C2T | (CH3)2CH- |
| GBB | -C=O-CH-NH- |
| ABB | -C=O-CH(CH3)NH- |
SPICA Force Field
Okayama University