SPICA: Surface Property fItting Coarse grAined model
Tutorialprotein
Step 1. Prepare initial configuration
We will introduce how to prepare an initial configuration of lipid-protein systems at an AA level. As seen in tutorial for lipid membrane systems, CHARMM-GUI is used to create AA PDB files of lipid-protein systems here. Since our tools are based on the PDB file format, we focus on how to prepare the intial coordinate files in a PDB format. The obtained PDB files will be used to generate CG PDB through a command of spica-tools map2cg. In the next step, to create a topology file of CG proteins in the CG PDB gained by mapping, we will apply a command of spica-tools ENM to a PDB file including only CG protein information. Then the whole CG PDB file generated by mapping will be converted to LAMMPS compatible input file using setup_lammps.AA PDB file: Initial configuration using CHARMM-GUI
CHARMM-GUI is a web tool, which generates initial coordinates for various systems. We use CHARMM-GUI to prepare initial coordinates of membrane protein system in an all-atom level.- Visit CHARMM-GUI web site and go to "Bilayer Builder" (Input Generator-Membrane Builder-Bilayer Builder)
- Scroll down and click "Protein/Membrane System"
- If you do not have protein PDB files for simulations, input PDBID of a protein you will use for "Dowload PDB File" and select "RCSB" or "OPM" for "Dowload Source".
If you have the protein PDB file for simulations, load the file for "Upload PDB File" and select "PDB Format".
For this turtial, we use a peripheral peptide (PDBID:1d6x) by downloading the PDB file from RCSB.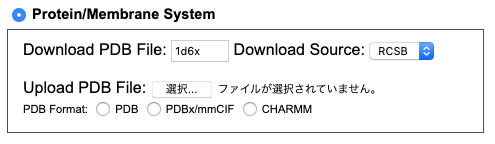
- Check protein models you want to contain in the initial configuration. PDB file for 1D6X has a single peptide. We select and click on one protein chain for simulations.
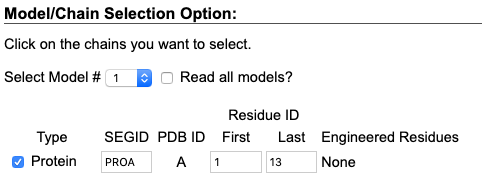
Then, Click "Next Step: Manipulate PDB". - Here we can manipulate the protein used to generate the initial configuration. For this tutorial, we focus on creating the configuration for CG-MD simulations; we do not manipulate the protein.

- Position and orientation of the protein model can be determined in this step. We want to simulate a peripheral peptide on a lipid membrane. We use "Positioning Options" to translate the peptide on the membrane surface.

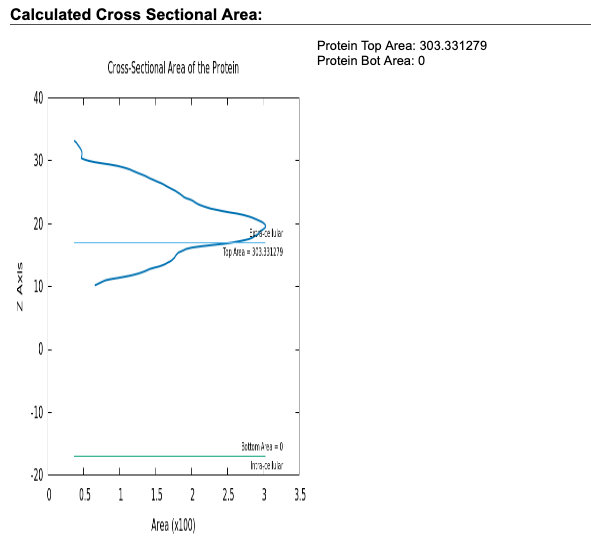
- Check the calulated cross-sectional area plotted in the web page of this step. The plot roughly shows z coordinate, normal to the lipid membrane, of proteins included in the configuration. In this tutorial, the plot shows that the peptide 1d6x is positioned around 20 A.
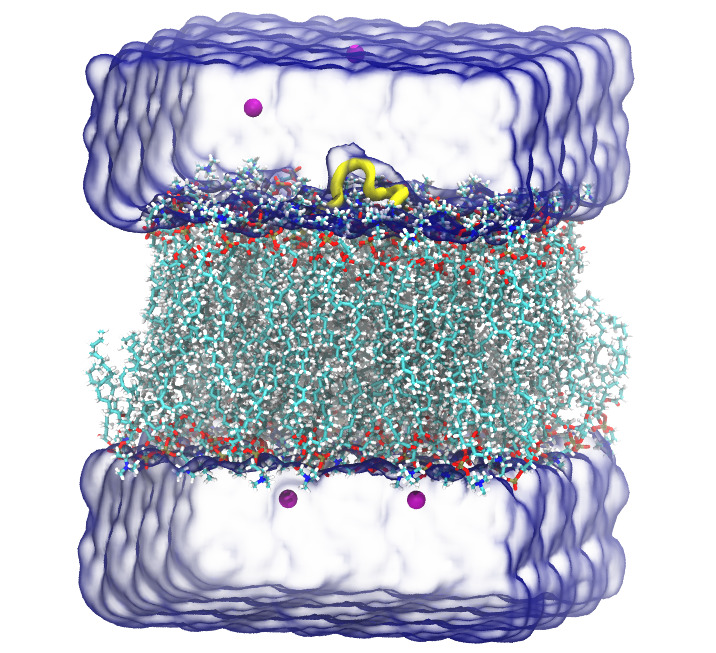
For this tutorial, we select "Rectangular" for "Box type", and input 50 for "Hydration #" and 64 for both "# of Lipid on Upperleaflet" and "# of Lipid on Lowerleaflet" of DOPC.
Since the peptide 1d6x has a net charge of +4, we add four chloride ions, as contour ion, to neutralize the system.
- After finishing all the remaining steps, as in Step 10 and 11 in tutorial of CHARMM-GUI for lipid membrane systems, one can find the files of the cooridnate("step5_assembly.pdb") and topology("step5_assembly.psf").
CG PDB file: Mapping AA to CG
After obtaining AA PDB file of the initial configuration, we map the AA PDB file to the CG PDB file using a command of spica-tools map2cg. For installation of the tools, please refer to the spica-tools GitHub repository here.To execute the mapping script, one should provide the input AA PDB file and the output CG PDB file name. The command will be:
$ cg_spica map2cg step5_assembly.pdb system.cg.pdb
where step5_assemply.pdb is the obtained AA PDB file using CHARMM-GUI and system.cg.pdb is the output CG PDB we named. The output file system.cg.pdb can be downloaded here for this tutorial.
| Previous | Top | Next |
SPICA Force Field
Research Institute for Interdisciplinary Science (RIIS)
Okayama University