SPICA: Surface Property fItting Coarse grAined model
Tutoriallipid membrane
Step 1. Prepare initial configuration
The initial configuration for MD simulation can be generated by various tools, such as packmol and VMD plugins. In this step, we show how to prepare an initial configuration of lipid membrane for SPICA-FF. There are two different ways to prepare the configurations:- Initial configuration at the CG resolution from the beginning.
- Prepare the initial configuration at the atomistic resolution and then map the configuration into the one at the CG resolution.
-
- To prepare a lipid membrane configuration with SPICA-FF,
packmol
could be most useful. For detailed instruction, one should refer to the packmol webpage,
though we here give an example of the packmol script useful for
a preparation of a DOPC membrane. Alternatively, you may want to start a simulation from
a random initial configuration. Packmol is still useful for this case,
also it may be useful for the preparation of a vesicle structure.
- MPDyn2
includes a tool to generate a random initial configuration for SPICA FF; GenCGconf.
Note that this program is not useful to prepare a lipid membrane.
This program will generate the initial configuration, initial.crd, and a PDB file, final.pdb.
The former file is useful for a CG-MD simulation using MPDyn2.
For lammps, the PDB file can be used as an input for the script, setup_lammps,
which will convert the configuration data into a data.file for lammps.
- To prepare a lipid membrane configuration with SPICA-FF,
packmol
could be most useful. For detailed instruction, one should refer to the packmol webpage,
though we here give an example of the packmol script useful for
a preparation of a DOPC membrane. Alternatively, you may want to start a simulation from
a random initial configuration. Packmol is still useful for this case,
also it may be useful for the preparation of a vesicle structure.
- To construct a model system of lipid membrane, CHARMM-GUI sholud be the most convenient webtool. Here, we briefly illustrate how we can generate a all-atom configuration using CHARMM-GUI. We use the PDB file after the step5 in the CHARMM-GUI input generator. In the next step, the obtained PDB file with the CHARMM all-atom description will be converted using a command of spica-tools.
Initial configuration of a DOPC bilayer using packmol
Packmol is a tool to place molecules in a simulation box with a desired geometry such as lipid bilayer and vesicle. A molecular structure of a single molecule should be given as an input. In this example, we make a DOPC bilayer in aqueous solution, for which we prepare the PDB files for a single DOPC molecule and a single CG water particle. A sample input script for packmol is attached here, which generate a membrane of 128 DOPC with 1300 water particles.A random configuration using CGgenConf (MPDyn2)
CGgenConf is a tool provided within MPDyn2 software package. This is useful to generate a random initial configurations at the CG level. There are two steps to make a random configuration within a simulation box.- Make a molecular library
- Pack the molecules picked up from the library
Make a molecular library
First, we need to generate a library of molecular structures for the molecules containing multiple beads. (not like water particle) Here we take a DOPC as an example to generate the molecular library.
A directory, ./MolLib should be made, where the library files are generated. Then, conduct ~/MPDyn2/bin/GenCGconf
Required information shoud be given interactively.
| # Do you want to make a library for a molecule? [Y or N] | |
| > Y | |
| # How many molecular structures do you (want to) have in the library? | |
| > 1000 | When you DON'T need the molecular library, put any POSITIVE number Arbitrary number should be written. For example, usually, the numbers within 1000-9999 should be enough. If it exceeds 10000, it gives error. |
| # Do you have have a reference structure for some components of the system? [Y or N] | |
| > N | Usually.... |
| # Name of PARAMETER file = ? | |
| > par_CG.prm | Put "D" for a default choice, "~/CGparam/par_CG.prm" |
| # Name of TOPOLOGY file = ? | |
| > top_CG.prm | Put "D" for a default choice, "~/CGparam/top_CG.prm" |
| # Number of molecular species = ? | |
| > 1 | We only make a library of DOPC, so here just answer 1. |
| # Molecular names ? Give them in one line. | |
| > DOPC | |
| # Number of Atoms in a Molecule ? Give them in one line. | |
| > 17 | In SPICA-CG, DOPC is made by 17 beads, so that put 17. |
Then, you will find 1000 configuration files in ./MolLib directory.
Pack the molecules picked up from the library
Next, we pack the generated library molecules in the simulation box. To do this, we again use GenCGconf. ~/MPDyn2/bin/GenCGconf
| # Do you want to make a library for a molecule? [Y or N] | |
| > N | This time, we use the previously generated library files |
| # How many molecular structures do you (want to) have in the library? | |
| > 1000 | This should be the number of molecular structures generated in the previous calculation. |
| # Do you have have a reference structure for some components of the system? [Y or N] | |
| > N | Again, usually put "N". If and only if you have a membrane structure beforehand, just put some water molecules in the simulation box, you may put "Y", but for now, simply put "N". |
| # Name of PARAMETER file = ? | |
| > par_CG.prm | Put "D" for a default choice, "~/CGparam/par_CG.prm" |
| # Name of TOPOLOGY file = ? | |
| > top_CG.prm | Put "D" for a default choice, "~/CGparam/top_CG.prm" |
| # Number of molecular species = ? | |
| > 2 | We want to generate the binary system of DOPC and water, and then put 2 |
| # Molecular names ? Give them in one line. | |
| > DOPC WAT | |
| # Number of Molecules ? Give them in one line. | |
| > 1296 3456 | |
| # Number of Atoms in a Molecule ? Give them in one line. | |
| > 17 1 | |
| # Periodic Boundary Condition is used or not? [Y or N] | |
| > Y | PBC is usually used, so that 'Y'. |
| # Density { rho [g/cm^3] } or the size of the simulation box{ Lx, Ly, Lz [A] }? | |
| > 1.0 | Giving a single value, it will be understood as density. Put the density [g/cm] or insert the cell size (Lx,Ly,Lz). If a sinble value is put, it is considered as a density value. Now put 1.0. |
This produces the random initial configuration in a cubic box; initial.crd and final.pdb. initial.crd is the file to be used in MPDyn. final.pdb is just a corresponding PDB file. The PDB file does not contain topology data, you may need a PSF file to visulize by vmd. A PSF file can be made using a tool, PSFCG, which is also available within MPDyn2 software package.
Membrane structure at all-atom detail made by CHARMM-GUI
CHARMM-GUI is a web tool, which generates initial coordinates for various systems. We often use CHARMM-GUI to prepare initial coordinates of lipid membrane system in all-atom detail (with CHARMM force field).- Visit CHARMM-GUI web site and go to "Bilayer Builder" (Input Generator-Membrane Builder-Bilayer Builder)
- Scroll down and click "Membrane Only System"
- Tick "Heterogeneous Lipid" and "Numbers of lipid components". Change other options if needed (i.e., Water thickness).
- For this tutorial, we will prepare DOPC lipid membrane. Expand "PC (phosphatidylcholine) Lipids". Input 64 for both "# of Lipid on Upperleaflet" and "# of Lipid on Lowerleaflet" of DOPC.
- Click "Show the system info" button. Then, "Calculated XY system Size" will appear.
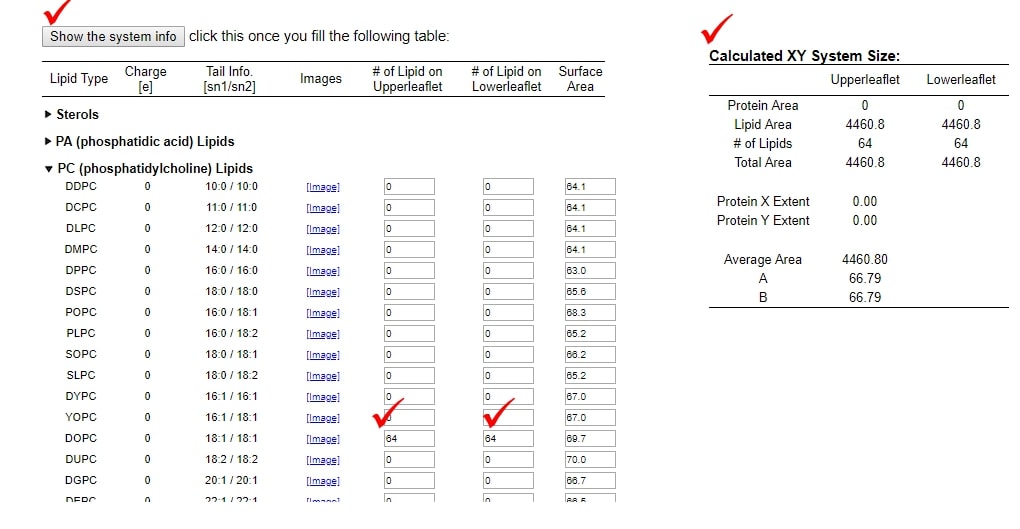
- Proceed to next step by clicking "Next Step: Determine the System Size"
- This step determines how many ions will be included in the system. Since PC lipids are neutral, we will not include ions in the system. Scroll down and untick "Include Ions". Then, go to next step.
- A message about the ions and water box will appear. Go to next step.
- CHARMM-GUI will assemble lipid membrane and solvation box. This step will take a while.
- The inital configuration is ready now. Download the files by clicking "download .tgz" in the information box.
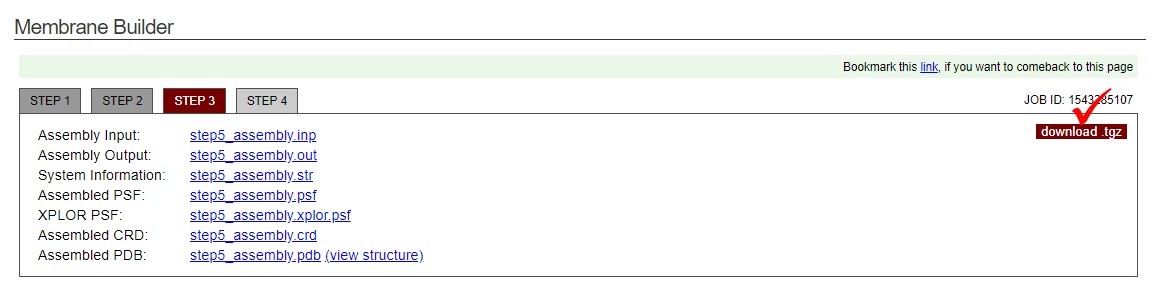
- Untar charmm-gui.tgz and one can find the files of the cooridnate("step5_assembly.pdb") and topology("step5_assembly.psf"). These files will be further converted to the LAMMPS compatible input file.
| Previous | Top | Next |
SPICA Force Field
Research Institute for Interdisciplinary Science (RIIS)
Okayama University